In a 2024 inspection of a mid-sized contract research organization, an FDA investigator asked the QA director to produce the version of a clinical trial protocol summary as it existed on a public study registry on a specific date eighteen months earlier. The CRO had the current version. It had a stack of meeting minutes that referenced earlier versions. What it did not have was the actual web record as it existed on the date the investigator asked about. The Form 483 that followed cited inadequate records of electronic content under 21 CFR Part 11.
21 CFR Part 11 is the FDA regulation that governs electronic records and electronic signatures used to satisfy any other FDA regulation. It applies to the pharmaceutical, biotechnology, medical device, food, and tobacco industries. It is the most-cited FDA recordkeeping regulation, and it is one of the most misunderstood. The phrase "Part 11 compliant" appears in every vendor pitch, but the underlying obligations cut across vendor selection, system validation, and the specific records the regulated firm decides to use as part of its quality system.
For most life sciences firms, the well-trodden Part 11 territory is laboratory information management systems, electronic batch records, and electronic CAPA systems, all of which are covered in detail by validation work. The territory that gets short shrift is web-based content: public-facing webpages, study registries, regulatory authority publications, vendor trust portals, and document repositories that the firm relies on to make GxP decisions. These are the records that fall through the cracks of a traditional validation program.
This guide covers what Part 11 requires for web-based records, when those records are in scope, how to build a Part 11 compliant web archive, the role of audit trails and timestamps, and how to set up the monitoring tooling without forcing every change through a formal validation effort.
What Part 11 Actually Requires
Part 11 has three sections that matter for web-based records: Subpart B (electronic records), Subpart C (electronic signatures), and the FDA's 2003 Guidance for Industry on the scope and application of Part 11.
Subpart B: electronic records
Section 11.10 requires controls for closed systems used to create, modify, maintain, or transmit electronic records. The list of controls is long but the substantive ones for web-based content include:
- Validation of systems to ensure accuracy, reliability, consistent intended performance, and the ability to discern invalid or altered records
- Generation of accurate and complete copies in human-readable and electronic form for inspection
- Protection of records to enable accurate and ready retrieval throughout the retention period
- Limiting system access to authorized individuals
- Use of secure, computer-generated, time-stamped audit trails to record the date and time of operator entries and actions that create, modify, or delete electronic records
The audit trail requirement is the single most-cited failure in Part 11 enforcement. A Part 11-compliant record without a tamper-evident audit trail of its creation and modification history is not, in fact, Part 11-compliant.
Subpart C: electronic signatures
Subpart C covers the binding of electronic signatures to records. Most web-based records that life sciences firms care about (regulatory authority pages, study registries, vendor portals) are not signed by the firm itself. They are read and relied on. Where electronic signatures attach to web records (electronic informed consent platforms, eTMF systems exposing approved documents through a web interface), Subpart C applies in full.
The 2003 Guidance: what FDA actually expects
The 2003 Guidance for Industry on Part 11 narrowed FDA's enforcement discretion materially. The agency stated that it would exercise enforcement discretion for some of the more onerous Part 11 requirements where firms apply a risk-based approach. In practice, this means:
- The firm decides which records are Part 11 records based on whether they are required by another FDA regulation
- The firm validates systems based on the risk those systems pose to product quality and patient safety
- Audit trails, copies of records, and record retention apply to all Part 11 records regardless
The risk-based approach does not eliminate the obligations. It allows the firm to scale the rigor to match the risk.
When Web-Based Records Are In Scope
A page on the public internet is not automatically a Part 11 record. It becomes a Part 11 record when the firm uses its content to satisfy a predicate FDA regulation. A few common scenarios where this applies:
Public study registries (clinicaltrials.gov, EudraCT/CTIS)
Posting a study to ClinicalTrials.gov is required under FDAAA 801. Information posted there is part of the regulatory record for the trial. If the firm relies on the registry posting (or its update) to satisfy a transparency obligation, the firm needs to retain a record of what was posted and when. Manual screenshots are not sufficient unless they are part of an audit-trail-bearing system.
FDA guidance and rulemaking
Quality system, GxP, and clinical trial design decisions are routinely justified by reference to FDA guidance documents (draft and final) and rulemakings. When a firm cites guidance to justify a deviation, study design, or quality decision, the version of the guidance as it existed at the time of the decision is part of the record. FDA guidance pages move; "withdrawn" guidance is removed without notice.
Vendor and supplier portals
GxP suppliers (CDMOs, CROs, lab service providers, analytical instrument vendors) publish quality certificates, ISO certifications, supplier qualification documents, and change notifications on web portals. When a firm relies on these to discharge its supplier qualification obligations, the portal pages become part of the firm's quality record.
Adverse event databases
FAERS, VAERS, EudraVigilance, and other public adverse event databases inform pharmacovigilance decision-making. When a firm's signal detection or risk evaluation cites these sources, retaining the data as it existed at the time of the analysis is critical.
Regulatory authority publications
EMA, MHRA, Health Canada, TGA, PMDA, and other authorities publish guidance, opinions, and approval decisions that inform GxP processes. Firms that operate across jurisdictions rely on these regularly.
Building a Part 11 Compliant Web Archive
The pattern that scales for life sciences QA, regulatory affairs, and pharmacovigilance teams is a continuous web monitoring system that captures content as it changes, produces tamper-evident archives, maintains the audit trail, and integrates with the firm's existing electronic records systems.
Identify the GxP-relevant URLs
Every regulatory affairs team has a working list of agency URLs they reference. Every pharmacovigilance team has a list of safety databases. Every QA team has a list of supplier portals. Every clinical operations team has a list of study registry URLs. Consolidating these into a single inventory, tagged by category and process owner, is the first step.
The categories that typically apply:
- Agency guidance pages (FDA, EMA, MHRA, Health Canada, TGA, PMDA)
- Compendial publications (USP, EP, JP)
- Supplier and CRO portals (vendor-specific)
- Public adverse event databases (FAERS, VAERS, EudraVigilance)
- Clinical trial registries (ClinicalTrials.gov, EudraCT/CTIS)
- Regulatory ICH publications
- Quality system supplier qualification pages
Set capture frequency by risk
GxP risk drives frequency. Agency safety advisories: hourly to daily. Adverse event database queries: continuous via scheduled checks. Supplier portal pages: daily for active suppliers, weekly for monitored-only. Compendial chapters: weekly. Study registry pages: daily for active studies during enrollment, weekly during follow-up.
The 2003 Guidance's risk-based approach applies here. Pages that affect immediate GxP decisions (safety advisories, supplier change notifications) merit high-frequency capture. Slower-moving guidance pages can be captured daily or weekly without raising risk.
Capture content with audit trail
For each detected change, the system must capture:
- Full HTML of the page
- Full screenshot
- Linked PDF documents
- Capture timestamp from a trusted source
- Cryptographic hash of the captured content
- An audit trail entry recording the capture event, the user or system that initiated it, and the linkage to the source URL
WACZ format archives include cryptographic hashes of every captured resource by default, which is well-suited to the Part 11 tamper-evidence requirement. Continuous monitoring systems can write a new WACZ entry for every detected change, building up an archive that is queryable by URL and date.
WACZ archive capture is available on request (contact us to enable audit-ready archiving for your account), set up per monitored page. On enabled pages, every detected change produces a WACZ archive sealed with a domain-identity signature using a Let's Encrypt certificate, an RFC 3161 timestamp from a commercial Trust Service Provider, and a Bitcoin blockchain anchor via OpenTimestamps. Each is independently verifiable with public tooling, and together they satisfy 21 CFR 11.10's audit-trail and tamper-evidence requirements without depending on a single provider's continued availability. For life sciences firms preparing for parallel EMA inspections, optional eIDAS qualified RFC 3161 timestamps from a QTSP on the EU Trusted List are available on Custom plans (Article 41 legal presumption).
The AI fabrication problem
In 2026 a generative model can produce a plausible screenshot of an FDA guidance page or a CRO portal notice in seconds. A self-stored screenshot proves nothing to an inspector because the firm could have generated it after the fact. What AI cannot fabricate is a hash anchored to the Bitcoin blockchain, an RFC 3161 timestamp signed by a Trust Service Provider's private key, or a qualified seal from a regulated QTSP. PageCrawl attaches several of these in parallel on every detected change. The archive's existence at a specific moment is attested by parties no single actor can spoof, which is the only practical bar for Part 11 records in an AI-saturated world.
Tie web records into the QMS
A Part 11 record that lives in a separate system from the firm's QMS is harder to retrieve, harder to audit, and easier to lose. The pattern that holds up under inspection is to treat the monitoring system as the capture layer and the QMS as the system of record. When a regulatory affairs analyst cites FDA guidance in a CAPA, the QMS record links to the captured archive entry that was current on the date of the CAPA.
This is not a heavy integration. It can be as simple as a link from the QMS record to the monitoring system's change-history page for the source URL. The captured archive entries on the monitoring system are tamper-evident and timestamped; the QMS record carries the link.
Validation expectations
Part 11 systems require validation. The level of rigor scales with risk per the 2003 Guidance. For a web monitoring and capture system used to retain externally-published records (not to author records the firm itself approves), the validation effort is typically lighter than for a fully-internal system. Common expectations:
- A user requirements specification identifying the records the system retains and the controls it provides
- An installation and operational qualification documenting that the system was installed correctly and operates as specified
- A performance qualification documenting that the system retains records consistent with the URS over a representative period
- A change-control procedure for changes to the system (vendor updates, frequency changes, scope expansion)
A monitoring vendor that produces consistent timestamps, tamper-evident archives, and a complete audit trail of capture events is materially easier to validate than a custom-built screenshot pipeline.
A Worked Example: Supplier Change Notification
A common pattern: a CDMO posts a notice on its quality portal that a critical reagent supplier has changed. The notice is dated and remains live for 60 days, after which it is archived to a secondary page that is not crawled by external search. The pharma firm relies on supplier change notifications to discharge its supplier qualification obligations under 21 CFR 211.84.
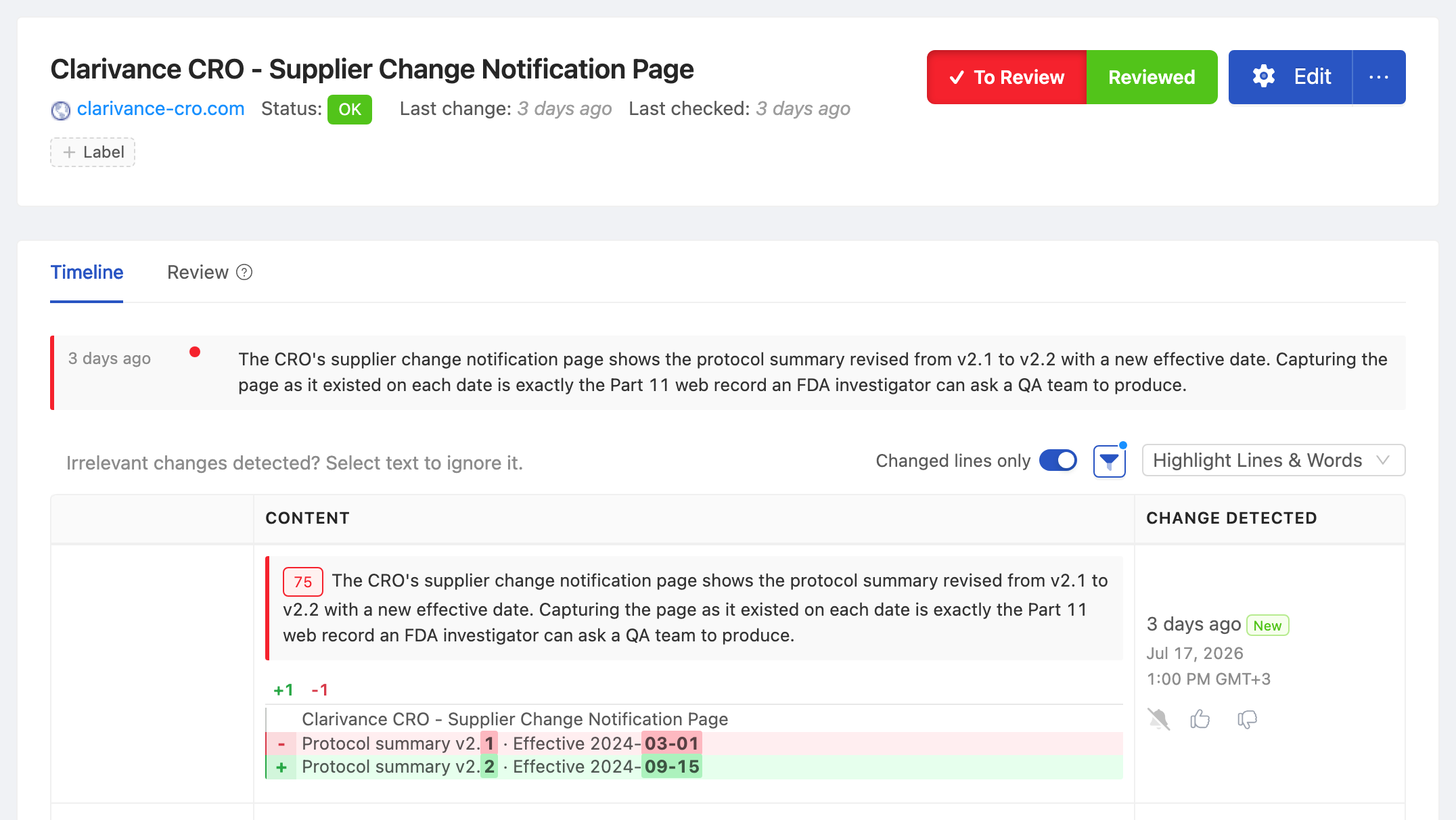
Without a continuous capture system, the firm has whatever screenshot the supplier qualification analyst happened to take. If the analyst was on PTO when the notice posted, or if the notice was archived before capture, the firm has no record of receipt.
With a continuous capture system, every change to the CDMO's quality portal is captured. The supplier change notification is archived with timestamp, full HTML, and any linked documents. The QMS record for the supplier qualification action links to the captured archive entry. When an FDA investigator asks for evidence that the firm received and acted on the notification, the linkage is intact.
Common Pitfalls
Treating screenshots as Part 11 records
A standalone screenshot is not a Part 11 record. It has no audit trail, no tamper-evidence, and no linkage to a capture system. Part 11 records require the audit-trail and integrity controls in 11.10.
Manual capture cadence
A weekly capture sweep misses changes between captures. For records that affect product quality or patient safety decisions, the gap is operationally significant. Continuous monitoring (capture on detected change) eliminates the gap.
Ignoring linked documents
Agency guidance pages link to PDF documents. CRO portals link to attached certificates. Adverse event databases link to case detail pages. The capture system has to follow links and retain the linked content as part of the same capture event, not as a separate ad-hoc step.
No risk classification
Capturing every page at the same frequency wastes effort on slow-moving pages and underweights fast-moving safety pages. A risk-based capture frequency, documented in the URS, satisfies the 2003 Guidance and produces a sustainable workload.
Treating the archive as cold storage
A capture system that only writes records and never lets the firm retrieve them is an audit-trail liability. Inspectors ask for retrieval. The archive needs to be queryable by URL, by date, and by tag. Retrieval is part of the system specification.
Choosing your PageCrawl plan
PageCrawl's Free plan lets you monitor 6 pages with 220 checks per month, which is enough to validate the approach on your most critical pages. Most teams graduate to a paid plan once they see the value.
| Plan | Price | Pages | Checks / month | Frequency |
|---|---|---|---|---|
| Free | $0 | 6 | 220 | every 60 min |
| Standard | $8/mo or $80/yr | 100 | 15,000 | every 15 min |
| Enterprise | $30/mo or $300/yr | 500 | 100,000 | every 5 min |
| Ultimate | $99/mo or $999/yr | 1,000 | 100,000 | every 2 min |
Annual billing saves two months across every paid tier. Enterprise and Ultimate scale up to 100x if you need thousands of pages or multi-team access.
Compliance monitoring is the cheapest insurance you can buy. A single missed regulatory change can trigger fines in the tens or hundreds of thousands, not to mention the audit overhead of proving you did not see it coming. Enterprise at $300/year covers 500 regulatory pages with unlimited history and timestamped screenshots, which is usually exactly what an assessor wants to see. All plans include the PageCrawl MCP Server, so your compliance team can ask Claude to summarize every change to a specific regulation over the last quarter and pull the exact diff, turning your monitoring history into a queryable audit trail. AI assistants can create monitors through conversation on every plan, including Free. Standard at $80/year is enough to cover 100 pages across your primary regulatory bodies if your program is smaller.
Getting Started
Set up Part 11 monitoring in three steps:
- Inventory your GxP-relevant URLs. Agency guidance pages, supplier portals, study registries, adverse event databases, compendia. Tag by category and process owner.
- Set risk-based capture frequency. Daily for safety advisories and active supplier portals. Weekly for guidance and slower-moving sources. Document the rationale.
- Document the system in your quality framework. URS, validation summary, audit-trail mechanism, retention period, retrieval procedure. Link captured records into the QMS where they support GxP decisions.
For related reading, see DORA compliance monitoring, clinical trial monitoring and FDA alerts, and SaaS sub-processor list monitoring.
If your life sciences team is building a Part 11 compliant web archive, the Regulatory Intelligence use case walks through the broader regulatory intelligence stack with workspace instructions, tag inheritance, and audit-grade timestamps that fit a validated quality system.